
Contents
Besides the devastating effects of the COVID-19 pandemic on human health and the global economy, it has also had a significant impact on the scientific community. Due to the enormity of the SARS-CoV-2 sequencing data that has been made publicly available, a hither-to unprecedented computational effort has to be made to analyse the data and infer globally meaningful conclusions comprehensively. Our group has been actively involved in creating novel databases to store and manage this large amount of information and developing novel computational pipelines and tools to analyze it.
While the GISAID database stores 16,475,549 SARS-CoV-2 consensus sequences, the raw sequencing datasets and systematic analysis results of more than 4,000,000 samples are available in the COVID-19 Data Portal.
The COVID-19 Data Portal
Our research group participates in The Versatile Emerging infectious disease Observatory project (VEO), which had a kick-off date of January 2020 and originally aimed to provide actionable information on a wide variety of emerging diseases and to promote early-warning using a versatile, interdisciplinary approach. However, with the rapid spread of COVID-19 starting from the end of 2019, much of the collaboration's initial efforts have naturally been directed towards SARS-CoV-2 monitoring and surveillance.
Among various achievements in the field, with the active support of the CSABAI•BIO Research Group, the consortium developed a standardised workflow to uniformly analyse raw SARS-CoV-2 next-generation sequencing data deposited in the European Nucleotide Archive (EBI-ENA) by an enormous number of researchers and institutions worldwide. A searchable database (European COVID-19 Data Portal) was created containing the mutations with their genomic positions, allele frequencies and additional metadata of the samples. The database is unique in the sense, that it contains the minor (low alternate allele frequency) variations in the samples in addition to the consensus sequences stored in GISAID.
The CoVEO database & CoVEO app
The systematic analysis results of the COVID-19 Data Portal have also been uploaded to the CoVEO PostgreSQL database, designed and maintained by our group. The database contains metadata, mutation calling results and information on sequencing quality and depth for all analysed samples. This detailed dataset allows for the identification of minor variants as well, which are not present in the consensus sequences of GISAID.
An overview of the samples stored in the CoVEO database can be obtained using the CoVEO app, which is also developed by our group. The explorer provides a user-friendly visualization interface to search for samples based on various metadata and variant profiles.
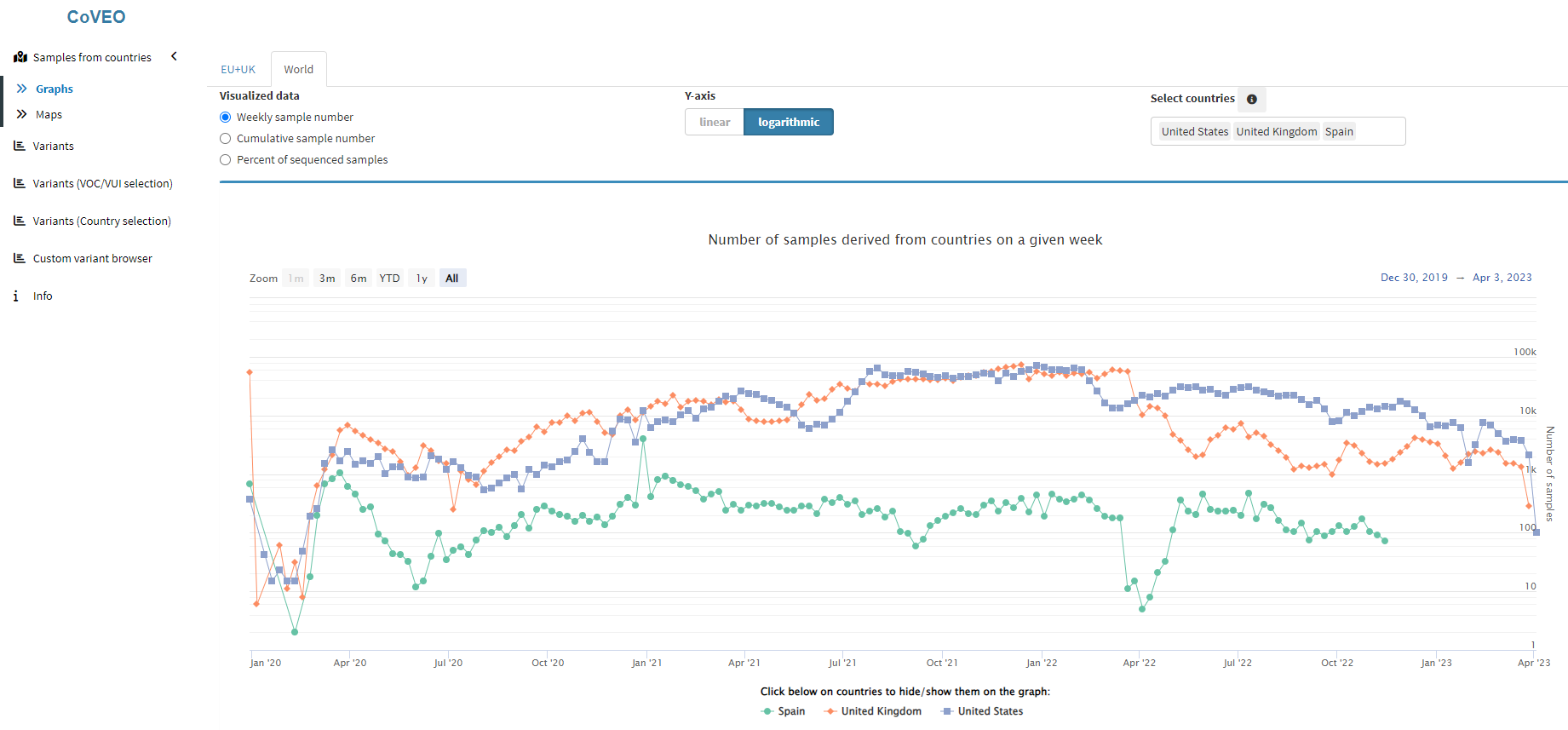
PCR primer efficiency & the ViralPrimer web server
The CoVEO database also contains information on the PCR primers used to amplify the SARS-CoV-2 genome in routine diagnostic tests. We used these primer sequences to identify mutations in the target regions of the commonly applied primers. Our approach distinguishes between mutations possibly having a damaging impact on PCR efficiency and ones anticipated to be neutral in this sense. Samples are categorized as "prone to misclassification" vs. "likely to be correctly detected" by a given PCR primer set based on the estimated effect of mutations present.
We found that samples susceptible to misclassification are generally present at a daily rate of 2% or lower, although particular primer sets seem to have compromised performance when detecting Omicron samples.
As different variant strains may temporarily gain dominance in the worldwide SARS-CoV-2 viral population, the efficiency of a particular PCR primer set may change over time, therefore constant monitoring of variations in primer target regions is highly recommended.
To this end, we developed the ViralPrimer interactive web application, which allows for the continuous evaluation of the efficiency of PCR primer sets against the currently dominant SARS-CoV-2 variants using either the dataset of GISAID or the CoVEO database. The application also provides the option to upload custom primer sequences to validate their performance against the available datasets of both SAR-CoV-2 and monkeypox sequences.
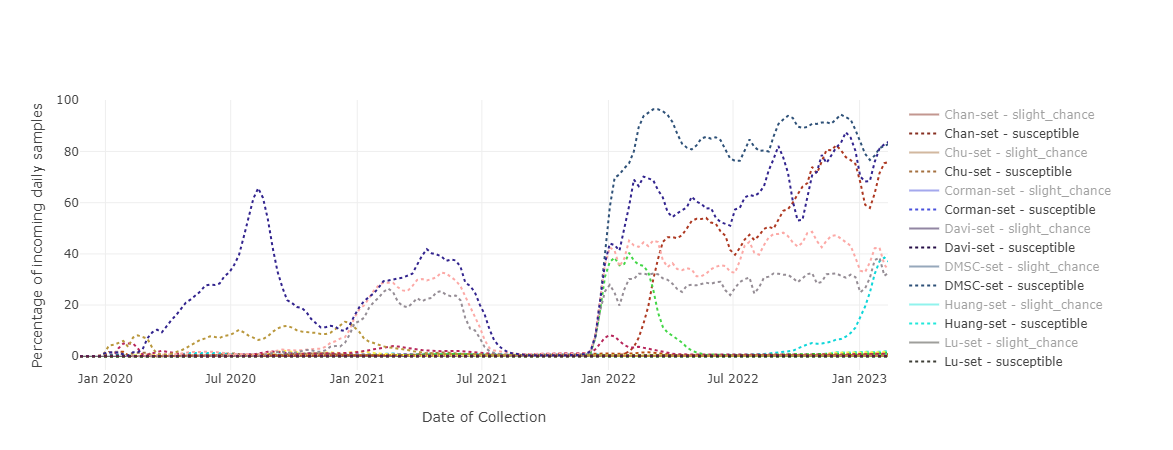
PCR primer sets that target a single region of the SARS-CoV-2 genome are especially vulnerable to mutations that affect the primer binding sites and that might be dominantly present in newly spreading variants. Whenever a significant portion of the circulating lineages carry such mutations (e.g. as a defining mutation), the efficiency of the particular primer set can drastically decrease, leading to false negative results.
Co-infection and intra-host recombination
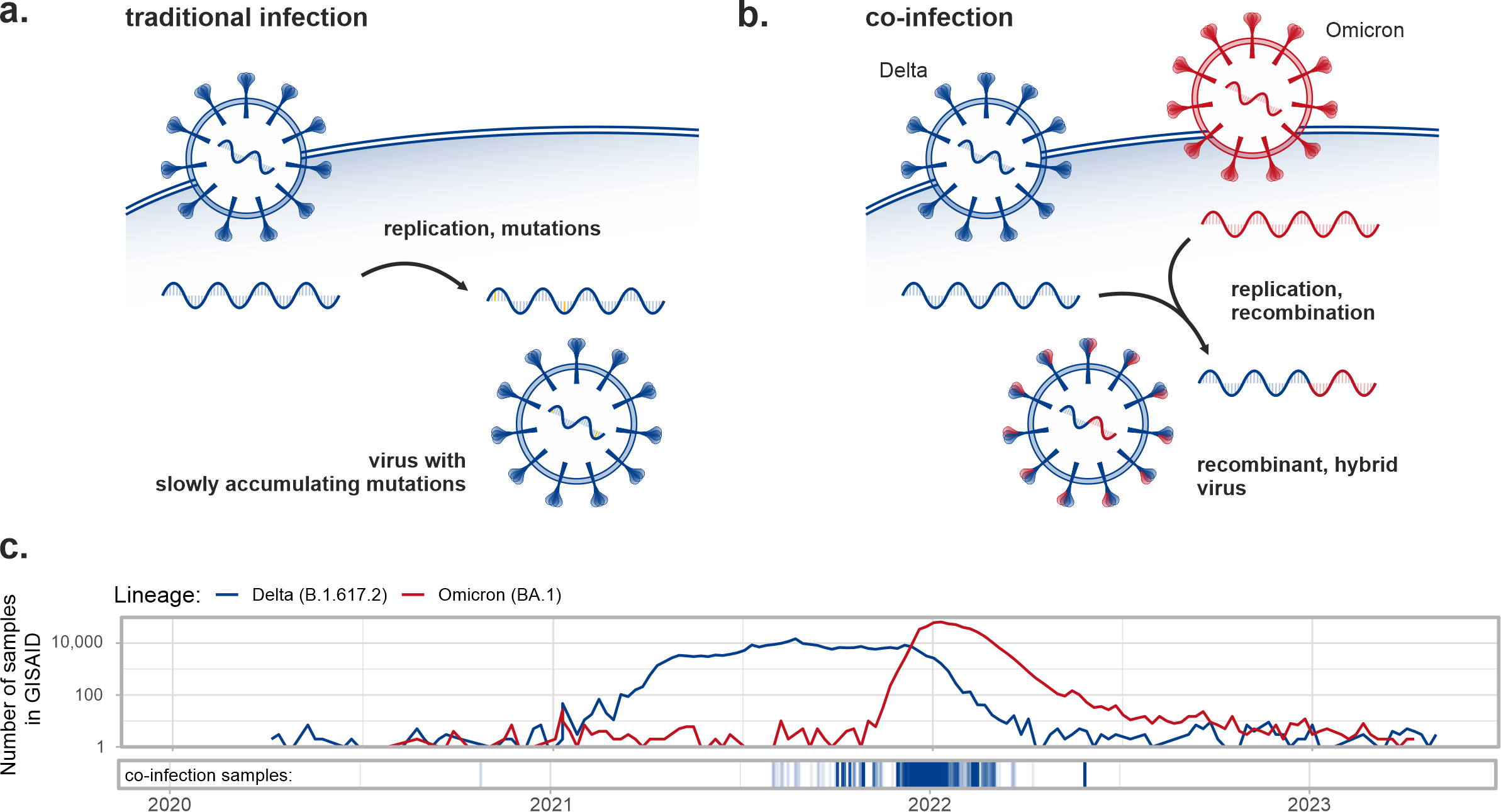
The CoVEO database also contains information on the possible presence of multiple viral strains in the same sample. This allows for the identification of co-infection events, which are of special interest in the case of SARS-CoV-2, as they may lead to the emergence of novel variants through intra-host recombination. We developed a computational pipeline to detect co-infection events in the CoVEO database and identified 7,700 samples (0.35% of all cases) with reliable evidence of multiple variants present, in most cases Delta-Omicron combinations.
Naturally, co-infections generally occurred in large numbers when the comprising variants were concurrently present and spreading in the investigated population.
If recombination occurs in a co-infection sample, traces of the recombinant genome might be detectable in a small fraction of the sequenced viral population, thus we further broadened our analysis to identify traces of intra-host recombination. However, the detection of such a scant genomic subpopulation is hampered by multiple factors. For instance, the distribution of defining mutations is not uniform along the genome, most of them are located on the S gene (coding the Spike protein), which makes it virtually impossible to detect recombination breakpoints in genomic regions, where the defining mutations of the two variants are scarce. Moreover, chimeric reads formed during the PCR amplification phase of amplicon sequencing are indistinguishable from real recombinant reads.
Keeping these sources of ambiguity in mind, we aimed to develop a pipeline, which can detect intra-host recombination in real-time, before the resulting recombinant genome spreads, for which we implemented two different approaches.
Deep mutational AlphaFold2 structures
In an attempt to bridge SARS-CoV-2 research with methods of AI, our team utilized the capabilities of AlphaFold2 (AF2) to compile an extensive dataset covering all mutations in the SARS-CoV-2 receptor-binding domain (RBD). Structural models were constructed for both the wild-type RBD and six variants, each featuring distinct amino acid mutations.
Comparing AF2-generated structures with experimental RBD-ACE2 complexes demonstrated excellent agreement, with an RMSD (Root Mean Square Deviation) of only 0.67 Angström.
We further employed phenotypic-genotypic models, utilizing different protein representations, including amino acid sequences, adjacency matrices, 3D structures, and bio-physical properties. UMAP dimensionality reduction revealed distinct clusters for different variants in both sequence and adjacency space, indicating structural similarities and dissimilarities.
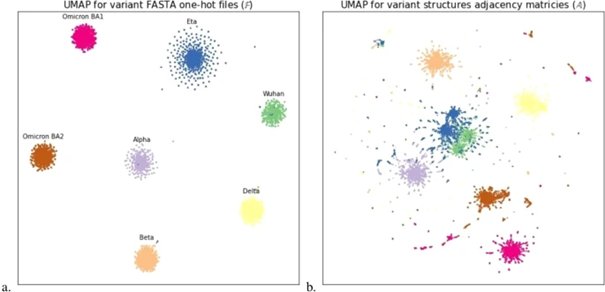
All AF2-generated models and related data are publicly accessible on FigShare.com in PDB or Fasta format.
Automated prediction of COVID-19 severity
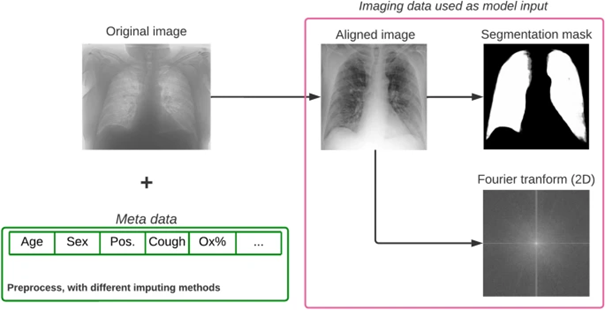
For another implementation of AI-based methods in COVID-19-related research, we took part in the "Covid CXR Hackathon—Artificial Intelligence for Covid-19 Prognosis" project, initiated in February, 2022 due to the significant challenges faced by Northern Italy during the early stages of the pandemic. The goal of the challenge was to develop an AI-based model capable of predicting the severity of COVID-19 upon admission based on chest X-ray (CXR) images and clinical metadata.
Through extensive analysis, we found that while CXRs provide valuable insights, they do not significantly enhance predictive power beyond the information available from other clinical data at admission.
This work highlights the potential of AI in improving medical prognosis by effectively combining different types of healthcare data.
Related publications
- Mentes et al. Identification of mutations in SARS-CoV-2 PCR primer regions. Sci Rep 12, 18651 (2022). DOI: 10.1038/s41598-022-21953-3
- Kilim et al. SARS-CoV-2 receptor-binding domain deep mutational AlphaFold2 structures. Sci Data 10, 134 (2023). DOI: 10.1038/s41597-023-02035-z
- Olar et al. Automated prediction of COVID-19 severity upon admission by chest X-ray images and clinical metadata aiming at accuracy and explainability. Sci Rep 13, 4226 (2023). DOI: 10.1038/s41598-023-30505-2
- Rahman et al. Mobilisation and analyses of publicly available SARS-CoV-2 data for pandemic responses. bioRXiv (2023). DOI: 10.1101/2023.04.19.537514
- Pipek et al. Systematic detection of co-infection and intra-host recombination in more than 2 million global SARS-CoV-2 samples. Nat Commun 15, 517 (2024). DOI: 10.1038/s41467-023-43391-z