
Contents
The biological process of aging is a complex phenomenon, which is influenced by a wide range of genetic, epigenetic, and environmental factors. The aging process is associated with a gradual decline in the functional capacity of cells, tissues, and organs, leading to an increased risk of age-related diseases and ultimately death. The identification of molecular markers of aging is of great interest, as it may provide insights into the underlying mechanisms of aging and age-related diseases, and may also have potential applications in the development of anti-aging interventions and personalized medicine.
In our research, we are particularly interested in the analysis of DNA methylation patterns, which are known to change with age and are considered to be one of the most promising molecular markers of aging. DNA methylation has been extensively used as a surrogate measure of biological age and correlations between “DNA methylation age” and chronological age have been established. A wide variety of epigenetic clocks has been designed to predict age in different tissues and on data obtained from different methylation platforms. We both use these existing clocks and develop our own refined versions for our explorations of the biological processes associated with aging.
A multi-tissue, multi-platform epigenetic clock
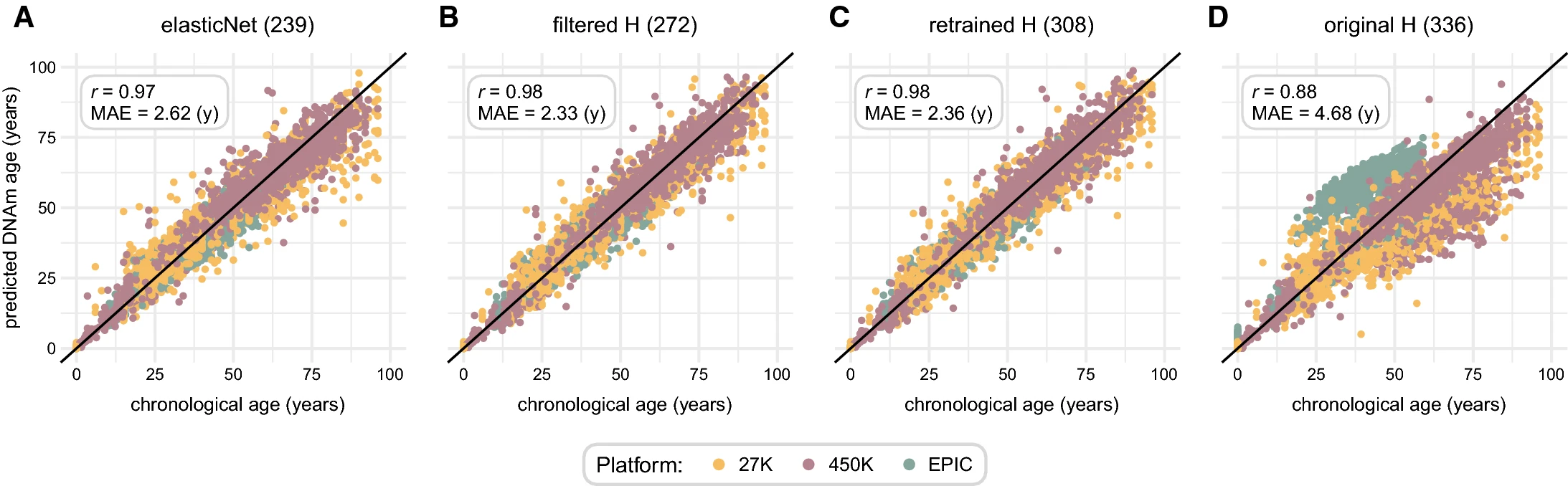
One of the most widely used epigenetic clocks is the Horvath pan-tissue clock, which was developed using methylation data from two different platforms (Illumina 27K and 450K arrays) and is able to predict chronological age in a wide range of tissues. However, the recent advent of the Illumina EPIC array, which covers a much larger portion of the genome, has raised the question of whether the Horvath clock can be applied to EPIC data without loss of accuracy. To address this question, we have developed a new multi-tissue, multi-platform epigenetic clock, which is based on a large dataset of methylation profiles from all Illumina platforms (27K, 450K, and EPIC).
We developed three models trained on close to 6,000 samples of various source tissues and platforms and demonstrate their superior performance (Pearson correlation (r) = 0.917-0.921 and median absolute error (MAE) = 3.60-3.85 years) compared to the original model (r = 0.880 and MAE = 5.13 years) on a test set of more than 4,000 samples.
The gain in accuracy was especially pronounced on EPIC array data (r = 0.89, MAE = 3.54 years vs. r = 0.83, MAE = 6.09 years), which was not available at the time when the original model was created.
Our updated epigenetic clocks predict chronological age with great precision in an independent test cohort of samples on multiple tissue types and data platforms. Two of the three presented models exclusively use the covariates of the original epigenetic clock, albeit with different coefficients, allowing for straightforward adaptation for prefiltered datasets previously processed with the original predictor.
Hierarchy and control of ageing-related methylation networks
Since the methylation of CpG dinucleotides function as switches in cellular mechanisms, it is plausible to assume that by proper adjustment of these switches age may be tuned. Though, adjusting hundreds of CpG methylation levels coherently may never be feasible and changing just a few positions may lead to biologically unstable state.
On the small subset of CpG dinucleotides defined by the Horvath pan-tissue epigenetic clock, we demonstrated how the adjustment of one methylation level leads to a cascade of changes at other sites. Among the studied subset, we located the most important CpGs (and related genes) that may have a large influence on the rest of the sub-system. According to our analysis, the structure of this network is way more hierarchical compared to what one would expect based on ensembles of uncorrelated connections. Therefore, only a handful of CpGs is enough to modify the system towards a desired state.
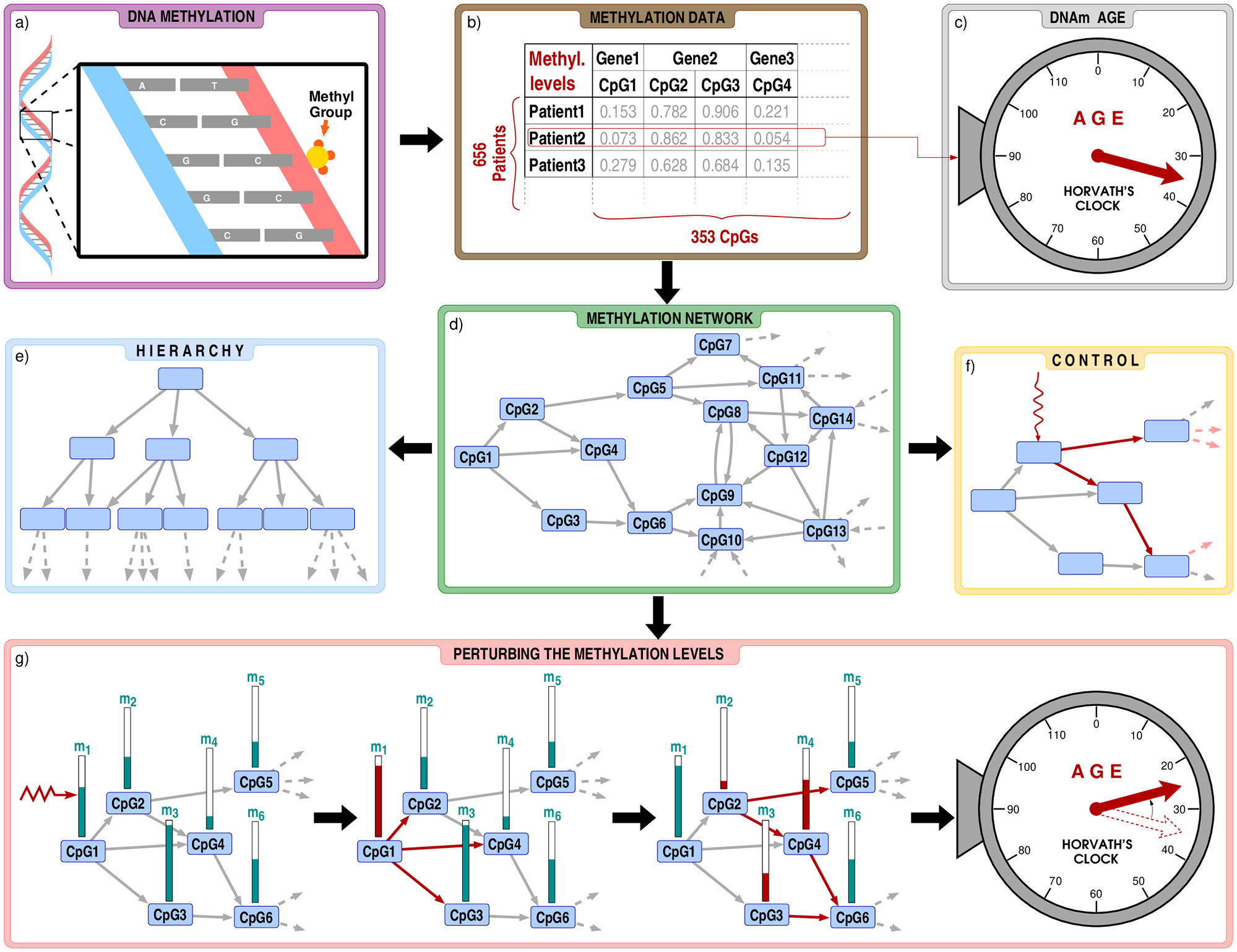
When propagation of the change over the network is taken into account, the resulting modification in the predicted age can be significantly larger compared to the effect of isolated CpG perturbations.
By adjusting the most influential single CpG site and following the propagation of methylation level changes we can reach up to 5.74 years in virtual age reduction, significantly larger than without taking into account of the network control.
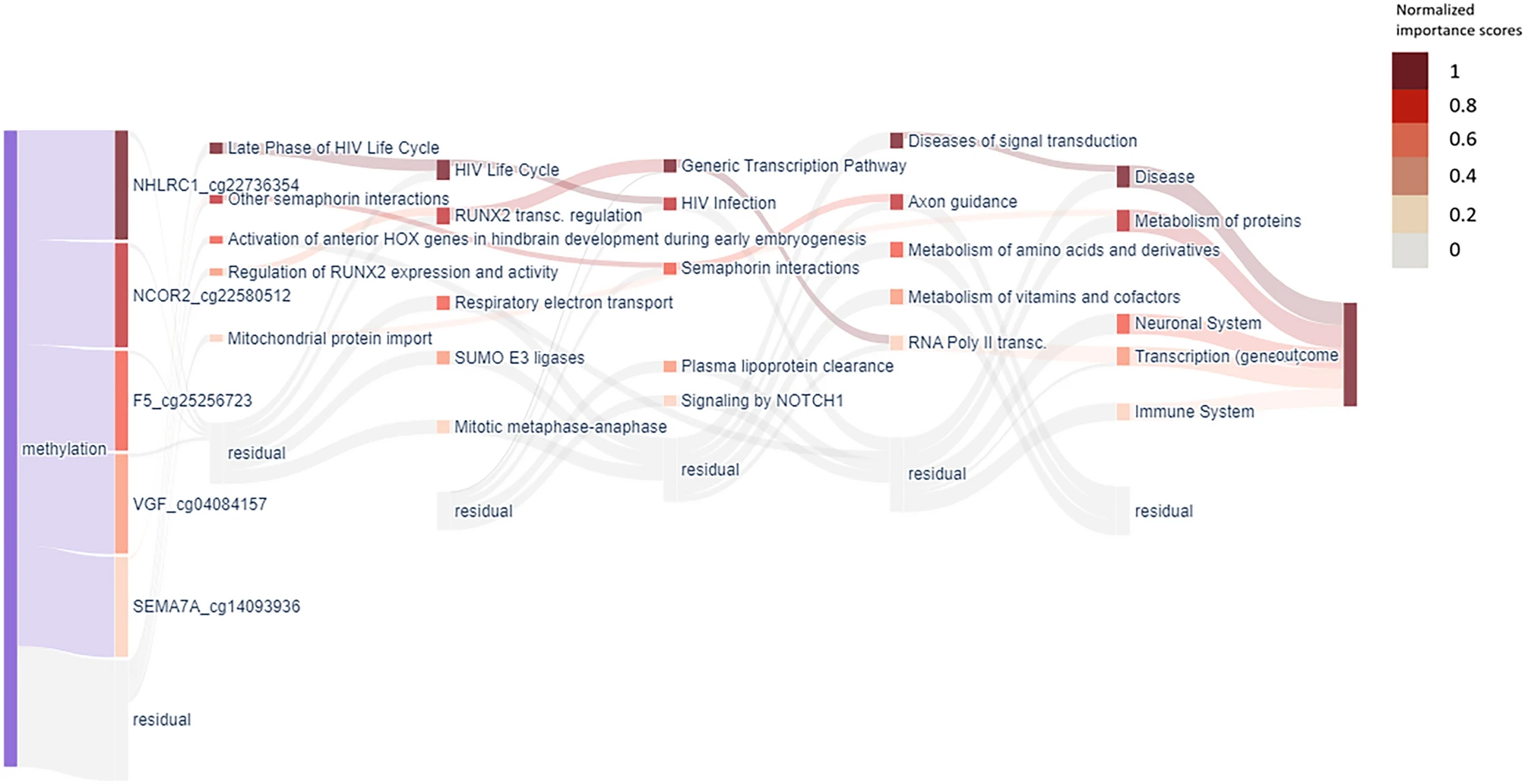
XAI-AGE: an explainable predictor of chronological age
Although epigenetic clocks can accurately estimate the biological age of an individual based on cellular DNA methylation, their models have limited ability to explain the prediction algorithm behind and underlying key biological processes controlling ageing. Our group has therefore developed XAI-AGE, a biologically informed, explainable deep neural network model for accurate biological age prediction across multiple tissue types.
We have shown that XAI-AGE outperforms the first-generation age predictors and achieves similar results to deep learning-based models, while opening up the possibility to infer biologically meaningful insights of the activity of pathways and other abstract biological processes directly from the model.
Related publications
- Pipek et al. A revised multi-tissue, multi-platform epigenetic clock model for methylation array data. Journal of Mathematical Chemistry 61 (2), 376-388 (2023). DOI: 10.1007/s10910-022-01381-4
- Palla et al. Hierarchy and control of ageing-related methylation networks. PLoS Comput Biol 17(9): e1009327 (2021). DOI: 10.1371/journal.pcbi.1009327
- Prosz et al. Biologically informed deep learning for explainable epigenetic clocks. Sci Rep 14, 1306 (2024). DOI: 10.1038/s41598-023-50495-5